EDIT:
This post is getting messier as I'm hammering things out...but I've gotten everything to work in the end, so please persist. The workflow described below is not the ideal one, but it'll get you started. I'll link here when I put up a newer, more reasonable tutorial.
EDIT2:
I'm really warming to ECCE as I'm learning more about it. I still think it'd be nice if it was open source, and I can't understand why it has to be reliant on csh (which is pretty much broken on ROCKS, and uncomfortable at the best of times), but it's pretty neat once you've got all the details ironed out. Error feedback/report could be better though.
EDIT 3:
ECCE is going open source the (northern) summer of 2012! As users we no longer have any excuses to complain.
Here's a quick introduction to getting started with using ECCE as the interface to nwchem, similar to how gaussview can be used to set up gaussian jobs.
This presumes that you've set up ECCE and preferably compiled your own version of nwchem:
http://verahill.blogspot.com.au/2012/03/ecce-on-debian-but-not-on-rockscentos.htmlhttp://verahill.blogspot.com.au/2012/03/nwchem-61-with-openmpi-on-rocks.htmlhttp://verahill.blogspot.com.au/2012/01/debian-testing-64-wheezy-nwhchem.html
##Important##Once I had figured all of this out I rebuilt nwchem and re-installed ecce in the proper locations. You might want to do the same.
A. If you're going to use several nodes you should put nwchem in the
same position in the file system hierarchy on all nodes e.g.
/opt/nwchem/nwchem-6.0/bin/LINUX64/nwchem
Also, make sure you share a folder (see how to use
NFS) between the nodes which you can use for run time files e.g.
/work
EDIT 4: This (probably) isn't necessary. In fact, using NFS in the wrong way will slow things down.
Set the permissions right (chown your user and set to 777 -- 755 is enough for nfs sharing between debian nodes, but between ROCKS and Debian you seem to need 777), and open your firewall on all ports for communication between the nodes.
B. Make sure that ECCE_HOME has been set in
~/.bashrc e.g.
export ECCE_HOME=/opt/ecce/appsand in
~/.cshrcsetenv ECCE_HOME=/opt/ecce/apps
C.edit
/opt/ecce/apps/siteconfig/submit.site (location depends on where you install ecce)
Change lines 65+ from
#NWChemCommand {
# $nwchem $infile > $outfile
#}
to (for multiple nodes)
NWChemCommand {
mpirun -hostfile /work/hosts.list -n $totalprocs --preload-binary /opt/nwchem/nwchem-6.0/bin/LINUX64/nwchem $infile > $outfile
}
to use mpirun for parallel job submissions and assuming you have a hosts file in /work. For running on a single node you can use
NWChemCommand {
mpirun -n $totalprocs $nwchem $infile > $outfile
}
user either
--preload-binary /opt/nwchem/nwchem-6.0/bin/LINUX64/nwchem or
$nwchem -- see what works for you. You probably can't do preload if you're running different linux distros (e.g. debian and centos)
My hosts.list looks like this:
tantalum slots=4 max_slots=4
beryllium slots=4 max_slots=5
Make sure that you don't accidentally put 2 jobs on node 0, then 2 jobs on node 1, then another 2 jobs on node 0, since they won't be consecutively numbered and will crash armci. You can avoid this by setting slots and max_slots to the same number.
D.You may have to edit
/etc/openmpi/openmpi-mca-params.conf if you have several (real or virtual) interfaces and add
e.g.btl=tcp,sm,self
btl_tcp_if_include=eth1,eth2
btl_tcp_if_exclude=eth0,virtbr0
Start ECCE:First start the server
csh /home/me/tmp/ecce/ecce-v6.2/server/ecce-utils/start_ecce_serverthen launch ecce
ecceThis will launch what the ecce people call the 'gateway':
 |
| The Gateway |
0. Make sure you've got your machine set upClick on Machine browser
 |
| Make sure that you can connect to the node e.g. by clicking on disk usage |
 |
| Set the application paths. Don't fiddle with nodes -- just change number of processors to the total for all nodes. |
1. Draw SiCl4 Click on the Builder in the Gateway, which gives you the following:
 |
| The builder window |
 |
| Click on More to get the periodic table which gives you access to Si |
 |
| Select Geometry -- here, Tetrahedral |
 |
| Si -- with four 'nubs' (yup, that's what the ecce ppl call them) |
 |
| Time to attach Cl atoms to the nubs. Select Cl and pick Terminal geometry. |
 |
| Click on a 'nub' to replace it with a Cl |
 |
| And do it until you've replaced all 'nubs'. Hold down right mouse button to rotate |
 |
| Click on the broom next to the bond menu on the right to pre-optimize the structure using MM |
 |
| And save. You will probably be limited to saving your jobs in folders below the ecce folder. |
2. Set up your jobClick on the Organizer icon in the 'gateway', which takes you here:
 |
| Click on the first icon, Editor |
 |
| Focus on selecting Theory and Run type. Here's we'll do a geometry optimisation. |
 |
| Click on Details for Theory |
 |
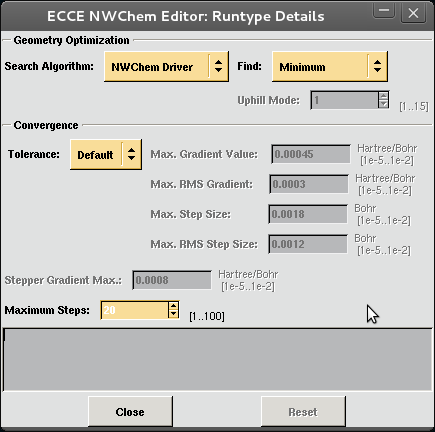
| Click on Details for Run type |
 |
| Constraints are optional |
 |
| In the organizer, click on the third icon to set the basis set. Defined atoms for a particular basis set are indicated by a n orange right lower corner |
 |
| You can get Details about the basis set |
 |
| If you don't have a Navy Triangle you can't run. Click on Editor and see what might be wrong. |
 |
| Ready to run. Click on Launch. |
4. Running |
| I'm still working on enabling more than a single core... |
Once you've clicked on launch you'll get
If you click on viewer you can monitor the job
 |
| Optimization in progress |
5. Re-launch a job at higher theoryIn the Organizer, select your last job and then click on Edit, Duplicate Setup with Last Geometry
 |
| You then get a copy to edit |
 |
| Change the basis set, save, then click on Final Edit |
 |
| This is the nwchem input file in a vim instance |
 |
| Add a line to the end, saying task scf freq to calculate the vibrations (there's another job option called geovib which does optim+freq , but here we do it by hand) |
 |
| Launch |
 |
| Running... |
 |
| You can now look at the vibrations |
 |
| And you can visualise MOs -- here's the HOMO which looks like all isolated p orbitals on the chlorine |
 |
| You can also calculate 'properties' |
 |
| These include GIAO shielding |
Performance:Here's phenol (scf/6-31g*) across three gigabit-linked nodes. The dotted line denotes node boundaries.
Here's a number of alkanes (scf/6-31g) on 4 cores on a single node:
This is dummy text. It is not meant to be read. Accordingly, it is difficult to figure out when to end it. But then, this is dummy text. It is not meant to be read. Period.
 If you click on viewer you can monitor the job
If you click on viewer you can monitor the job




































ConversionConversion EmoticonEmoticon