Update 2: The coordinates are actually gradients, and so aren't terribly informative to a casual user like myself. See this post for how to extract the geometries properly: http://verahill.blogspot.com.au/2013/08/506-extracting-optimized-structures.html
Update:
Please note that the coordinates in square brackets ([]) in the python output are not raw coordinates for the atoms in the molecule -- I haven't quite figured out how they scale, but it's not a simple matter of just multiplying. The energies are good though, and you can always extract the coordinates the slow and painful way by manually going through the output.
Another issue which should be stressed is that scan_input(geom,[1.398],[3.398],19,'dft',task_optimize) does not do the end points -- i.e. you won't get the energy for a bond length of 1.398, and you won't get the energy for a bond length of 3.398. Instead you'll get 19 data points in between these. It's a bit...awkward.
Original post:
A long time ago I made a post on doing potential energy surface (PES) scans in nwchem using python.
This is a post giving PES another look. The impetus for the post is that I'm tired of Gaussian failing and being opaque about the whole procedure.
The following page was of great help: http://www.fqt.izt.uam.mx/html/software_fqt/user/node34.html
NOTE: you'll need to compile nwchem with python support. See e.g. http://verahill.blogspot.com.au/2013/06/449-nwchem-63-updated-sources-compiling.html (the post is a bit messy, but persevere -- it's not that difficult)
On Debian the key is to change
in config/makefile.h before compiling. It's not necessary on RHEL clones.
Below I'll show three examples:
* a simple bond dissociation reaction. I also discuss the use of 'constant', and task_energy vs task_optimize.
* an SN2 reaction (CH3Br + I-)
* a 2D/parallel PES scan of ethane ( C-C bond length, H-C-C angle). I also show constant vs free variables.
Example 1.
Breaking the C-O bond in methanol
I set this up in ecce (see e.g. next example), but you don't have to. The input file I used was the following:
Also note that a constrained (i.e. not relaxed) PES scan can be done by doing task_energy instead of task_optimize.
ECCE can't quite handle the textual output (alt+O) since there are lines that are too long. The output is properly written though -- you'll just have to look in the Output folder of the job. The ecce.out file works fine though.
The job takes 90-100 seconds on an old 3-core node (AMD Athlon II X3).

The very end of the output file has all the results, but in a non-obvious way:
where A is the first step, S is the 19th step etc. This way you get 19x3+1=58 lines. This is clearly idiotic.
Instead, you can look through the output and search for 'Scanning NWChem input - results from step' to see all the output for the optimised structures one by one:
and then plot it:

Example 2.
SN2 reaction between iodide and bromomethane
You can set up your calc however you want, but ECCE is easier than anything else.
Draw bromomethane, then throw in an iodine atom. Adjust the angle across Br-C-I to 180 degrees, and set the C to I distance to 3 Å.

Set up the calculation -- in this case I used b3lyp/def2-svp
 Edit the input and add
Edit the input and add
(Delete 'task dft optimize')

You'll now have the following input file:

Example 3:
Two-dimensional PES scan
I'll keep this brief. First we do a scan where we use 'constant' for the angle, but not the bond length:
And the output:


If we instead set constant for the bond as well:
And we get:


Update:
Please note that the coordinates in square brackets ([]) in the python output are not raw coordinates for the atoms in the molecule -- I haven't quite figured out how they scale, but it's not a simple matter of just multiplying. The energies are good though, and you can always extract the coordinates the slow and painful way by manually going through the output.
Another issue which should be stressed is that scan_input(geom,[1.398],[3.398],19,'dft',task_optimize) does not do the end points -- i.e. you won't get the energy for a bond length of 1.398, and you won't get the energy for a bond length of 3.398. Instead you'll get 19 data points in between these. It's a bit...awkward.
Original post:
A long time ago I made a post on doing potential energy surface (PES) scans in nwchem using python.
This is a post giving PES another look. The impetus for the post is that I'm tired of Gaussian failing and being opaque about the whole procedure.
The following page was of great help: http://www.fqt.izt.uam.mx/html/software_fqt/user/node34.html
NOTE: you'll need to compile nwchem with python support. See e.g. http://verahill.blogspot.com.au/2013/06/449-nwchem-63-updated-sources-compiling.html (the post is a bit messy, but persevere -- it's not that difficult)
On Debian the key is to change
EXTRA_LIBS += -lnwcutil -lpthread -lutil -ldlto
EXTRA_LIBS += -lnwcutil -lpthread -lutil -ldl -lssl -lz
in config/makefile.h before compiling. It's not necessary on RHEL clones.
Below I'll show three examples:
* a simple bond dissociation reaction. I also discuss the use of 'constant', and task_energy vs task_optimize.
* an SN2 reaction (CH3Br + I-)
* a 2D/parallel PES scan of ethane ( C-C bond length, H-C-C angle). I also show constant vs free variables.
Example 1.
Breaking the C-O bond in methanol
I set this up in ecce (see e.g. next example), but you don't have to. The input file I used was the following:
The PES bit is highlighted in blue. Note the 'constant' keyword -- if you omit that the bond length will initially be set to whatever you define it to in your scan, but it can relax back to the optimum length. If you DO set 'constant' everything BUT that bond will be relaxed. Most likely this is what you will want to do.
scratch_dir /scratch
Title "meoh_pes"
Start meoh_pes
echo
charge 0
geometry autosym units angstrom
C 0.0351714 0.00548884 0.0351714
H -0.617781 -0.634073 0.667983
H 0.667983 -0.634073 -0.617781
H -0.605139 0.646470 -0.605139
O 0.839603 0.818768 0.839603
H 1.38912 0.201564 1.38912
end
ecce_print ecce.out
basis "ao basis" cartesian print
H library "3-21G"
O library "3-21G"
C library "3-21G"
END
dft
mult 1
direct
XC b3lyp
grid fine
iterations 99
mulliken
end
driver
default
maxiter 888
end
python
from nwgeom import *
geom = '''
geometry adjust
zcoord
bond 1 5 %f cccc constant
end
end
'''
results=scan_input(geom,[1.398],[3.398],19,'dft',task_optimize)
for i in range(0,len(results)):
print results[i][0][0],results[i][1]
end
task python
Also note that a constrained (i.e. not relaxed) PES scan can be done by doing task_energy instead of task_optimize.
ECCE can't quite handle the textual output (alt+O) since there are lines that are too long. The output is properly written though -- you'll just have to look in the Output folder of the job. The ecce.out file works fine though.
The job takes 90-100 seconds on an old 3-core node (AMD Athlon II X3).

The very end of the output file has all the results, but in a non-obvious way:
All in all, there are 58 lines for 19 steps. I think that there are three things happening -- firstly, the line in blue is the output from the 19th step, and that somehow gets mixed in with the output from all the calculations. Secondly, the structure and energy of each step is reported twice at a time. Thirdly, the optimised structures/energies are reported one more time by injecting them into the output, like this:
1.498 (-115.07289914310056, [-0.00130778291169336, 0.01798903956433226, 0.0, -4.009155466250247e-05, 1.693340302064139e-05, -6.637550254401381e-06, -4.009155466250247e-05, 1.693340302064139e-05, 6.637550254401381e-06, 2. 4514244186701895e-05, -1.5885649893555842e-05, 0.0, 0.0012636893525275195, -0.018041103298149008, 0.0, 9.97624242821682e-05, 3.4082577691996185e-05, 0.0])
(-114.8737952986994, [-4.7287277448850376e-05, 0.030029200359777717, 0.0, -1.3711175166353229e-06, -8.452926738775068e-08, 9.941241931599176e-07, -1.3711175166353229e-06, -8.452926738775068e-08, -9.941241931599176e-07, 8. 167348279908282e-07, -2.5820569179275075e-06, 0.0, 4.871429991895604e-05, -0.030027845123621805, 0.0, 4.984777179639632e-07, 1.3958792967685985e-06, 0.0])
1.498 (-115.07289914310056, [-0.00130778291169336, 0.01798903956433226, 0.0, -4.009155466250247e-05, 1.693340302064139e-05, -6.637550254401381e-06, -4.009155466250247e-05, 1.693340302064139e-05, 6.637550254401381e-06, 2. 4514244186701895e-05, -1.5885649893555842e-05, 0.0, 0.0012636893525275195, -0.018041103298149008, 0.0,
[..]
3.198 (-114.87977711993531, [-0.00018979360652668711, 0.033296276783081655, 0.0, -2.3787379704320877e-06, 1.7510009376556918e-06, 1.3530564600128248e-06, -2.3787379704320877e-06, 1.7510009376556918e-06, -1.3530564600128248e-06, 8. 24207064487048e-06, -8.055936327900498e-07, 0.0, 0.00018027576986845428, -0.03329589479259992, 0.0, 6.033241931824307e-06, -3.0783987173960137e-06, 0.0])
3.298 (-114.8737952986994, [-4.7287277448850376e-05, 0.030029200359777717, 0.0, -1.3711175166353229e-06, -8.452926738775068e-08, 9.941241931599176e-07, -1.3711175166353229e-06, -8.452926738775068e-08, -9.941241931599176e-07, 8. 167348279908282e-07, -2.5820569179275075e-06, 0.0, 4.871429991895604e-05, -0.030027845123621805, 0.0, 4.984777179639632e-07, 1.3958792967685985e-06, 0.0])
AS
A
B
B
C
C
D
D
A
E
E
B
where A is the first step, S is the 19th step etc. This way you get 19x3+1=58 lines. This is clearly idiotic.
Instead, you can look through the output and search for 'Scanning NWChem input - results from step' to see all the output for the optimised structures one by one:
In this particular case I can grep my way through by doing
Scanning NWChem input - results from step 2
(-115.06618436935011, [-0.0038228970733096973, 0.050051062094932305, 0.0, 2.9196769046224702e-05, -6.928661348853948e-06, 4.746536668570611e-06, 2.9196769046224702e-05, -6.928661348853948e-06, -4.746536668570611e-06, -1.0103262985700079e-05, 1.6491089715894858e-05, 0.0, 0.003767244388907326, -0.05005618579508188, 0.0, 7.362409274846993e-06, 2.489933151654522e-06, 0.0])
cat nwch.nwout |grep '^(-'|cat -nNot pretty, but manageable.
1 (-115.07289914310056, [-0.00130778291169336, 0.01798903956433226, 0.0, -4.009155466250247e-05, 1.693340302064139e-05, -6.637550254401381e-06, -4.009155466250247e-05, 1.693340302064139e-05, 6.637550254401381e-06, 2.4514244186701895e-05, -1.5885649893555842e-05, 0.0, 0.0012636893525275195, -0.018041103298149008, 0.0, 9.97624242821682e-05, 3.4082577691996185e-05, 0.0])
2 (-115.06618436935011, [-0.0038228970733096973, 0.050051062094932305, 0.0, 2.9196769046224702e-05, -6.928661348853948e-06, 4.746536668570611e-06, 2.9196769046224702e-05, -6.928661348853948e-06, -4.746536668570611e-06, -1.0103262985700079e-05, 1.6491089715894858e-05, 0.0, 0.003767244388907326, -0.05005618579508188, 0.0, 7.362409274846993e-06, 2.489933151654522e-06, 0.0])
3 (-115.05478103866017, [-0.005033784212299788, 0.06848598587431667, 0.0, -1.3396548676491982e-06, -2.5875637174599397e-08, -5.261746410523127e-07, -1.3396548676491982e-06, -2.5875637174599397e-08, 5.261746410523127e-07, 1.4459720645843e-07, -2.8328952926398587e-06, 0.0, 0.005034455335082233, -0.0684825786855032, 0.0, 1.8635897582608418e-06, -5.225422206114883e-07, 0.0])
4 (-115.04079235517, [-0.005485543277166251, 0.07798880362126945, 0.0, 4.745460307237215e-06, -5.597510268573469e-06, 5.645418744981701e-07, 4.745460307237215e-06, -5.597510268573469e-06, -5.645418744981701e-07, -6.651712157745848e-07, 6.750842351778419e-06, 0.0, 0.00548062073181968, -0.07798086728839469, 0.0, -3.903204054994669e-06, -3.4921546817404114e-06, 0.0])
5 (-115.02560006674966, [-0.0054233976595857575, 0.08166232318137269, 0.0, -1.659239761503395e-06, -4.376603580866223e-07, 4.4580035316599265e-06, -1.659239761503395e-06, -4.376603580866223e-07, -4.4580035316599265e-06, 3.034808945895362e-06, -6.726118036586015e-06, 0.0, 0.005436665955901393, -0.08164730868562775, 0.0, -1.2984625724410392e-05, -7.4130570159938736e-06, 0.0])
[..]
16 (-114.89364787840326, [-0.0005591249462735259, 0.04018795560035916, 0.0, -5.34666220519675e-07, 1.1370871814235517e-06, 4.809133242467123e-07, -5.34666220519675e-07, 1.1370871814235517e-06, -4.809133242467123e-07, -6.9140095421138525e-06, -3.095664552260277e-06, 0.0, 0.0005695756951453745, -0.040185884820554796, 0.0, -2.467406898132296e-06, -1.2492896190128416e-06, 0.0])
17 (-114.8863872514371, [-0.00036666056940981573, 0.03667976502852128, 0.0, 2.9101399354747315e-06, -2.094045026924257e-06, -4.933288234976185e-06, 2.9101399354747315e-06, -2.094045026924257e-06, 4.933288234976185e-06, 1.6531622304416516e-07, 1.511517903679191e-07, 0.0, 0.00036162347288279384, -0.03668602744257765, 0.0, -9.484995716624312e-07, 1.0299352320775057e-05, 0.0])
18 (-114.87977711993531, [-0.00018979360652668711, 0.033296276783081655, 0.0, -2.3787379704320877e-06, 1.7510009376556918e-06, 1.3530564600128248e-06, -2.3787379704320877e-06, 1.7510009376556918e-06, -1.3530564600128248e-06, 8.24207064487048e-06, -8.055936327900498e-07, 0.0, 0.00018027576986845428, -0.03329589479259992, 0.0, 6.033241931824307e-06, -3.0783987173960137e-06, 0.0])
19 (-114.8737952986994, [-4.7287277448850376e-05, 0.030029200359777717, 0.0, -1.3711175166353229e-06, -8.452926738775068e-08, 9.941241931599176e-07, -1.3711175166353229e-06, -8.452926738775068e-08, -9.941241931599176e-07, 8.167348279908282e-07, -2.5820569179275075e-06, 0.0, 4.871429991895604e-05, -0.030027845123621805, 0.0, 4.984777179639632e-07, 1.3958792967685985e-06, 0.0])
cat nwch.nwout |grep '^(-'|sed 's/\,/\t/g;s/(\([^)]*\))/\1/g'|cat -n|gawk '{print $1,$2}' > profile.dat
and then plot it:

Example 2.
SN2 reaction between iodide and bromomethane
You can set up your calc however you want, but ECCE is easier than anything else.
Draw bromomethane, then throw in an iodine atom. Adjust the angle across Br-C-I to 180 degrees, and set the C to I distance to 3 Å.


python
from nwgeom import *
geom = '''
geometry adjust
zcoord
bond 1 6 %f cccc constant
end
end
'''
results=scan_input(geom,[3.00],[1.5],20,'dft',task_optimize)
for i in range(0,len(results)):
print results[i][0][0],results[i][1]
end
task python
(Delete 'task dft optimize')

You'll now have the following input file:
Launch it and wait...eventually (2h 30 min on a slow three-core node) you'll get an output like the one below. Note that I didn't pre-optimise the bromomethane, so there's a bit of a drop in energy at the beginning. Likewise, I let the C-I distance get so short that the energy is rising rapidly at the endscratch_dir /scratch
Title "sn2_br"
Start sn2_br
echo
charge -1
geometry noautosym units angstrom
C 0.00000 0.00000 0.00000
H -0.675500 -0.675500 0.675500
H 0.675500 -0.675500 -0.675500
H -0.675500 0.675500 -0.675500
Br 1.10274 1.10274 1.10274
I -1.73205 -1.73205 -1.73205
end
ecce_print ecce.out
basis "ao basis" spherical print
H library "def2-svpd"
Br library "def2-svpd"
C library "def2-svpd"
I library "def2-svpd"
END
ECP
I library "def2-ecp"
END
dft
mult 1
direct
XC b3lyp
grid fine
iterations 99
mulliken
end
driver
default
maxiter 99
end
python
from nwgeom import *
geom = '''
geometry adjust
zcoord
bond 1 6 %f cccc constant
end
end
'''
results=scan_input(geom,[3.00],[1.5],20,'dft',task_optimize)
for i in range(0,len(results)):
print results[i][0][0],results[i][1]
end
task python
 |
| Structure at the beginning |
 |
| Transition-state-ish structure |
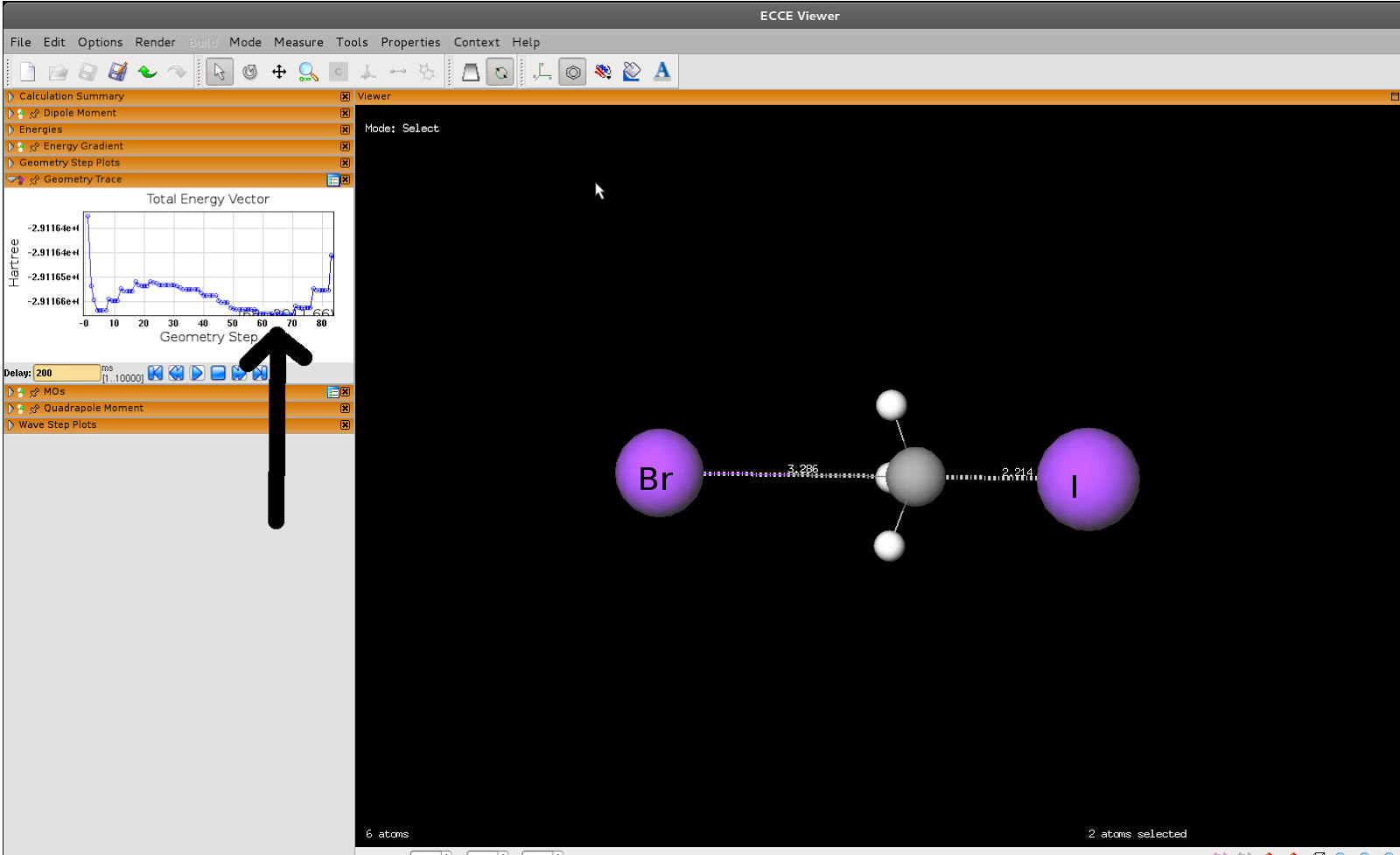 |
| Product |

Example 3:
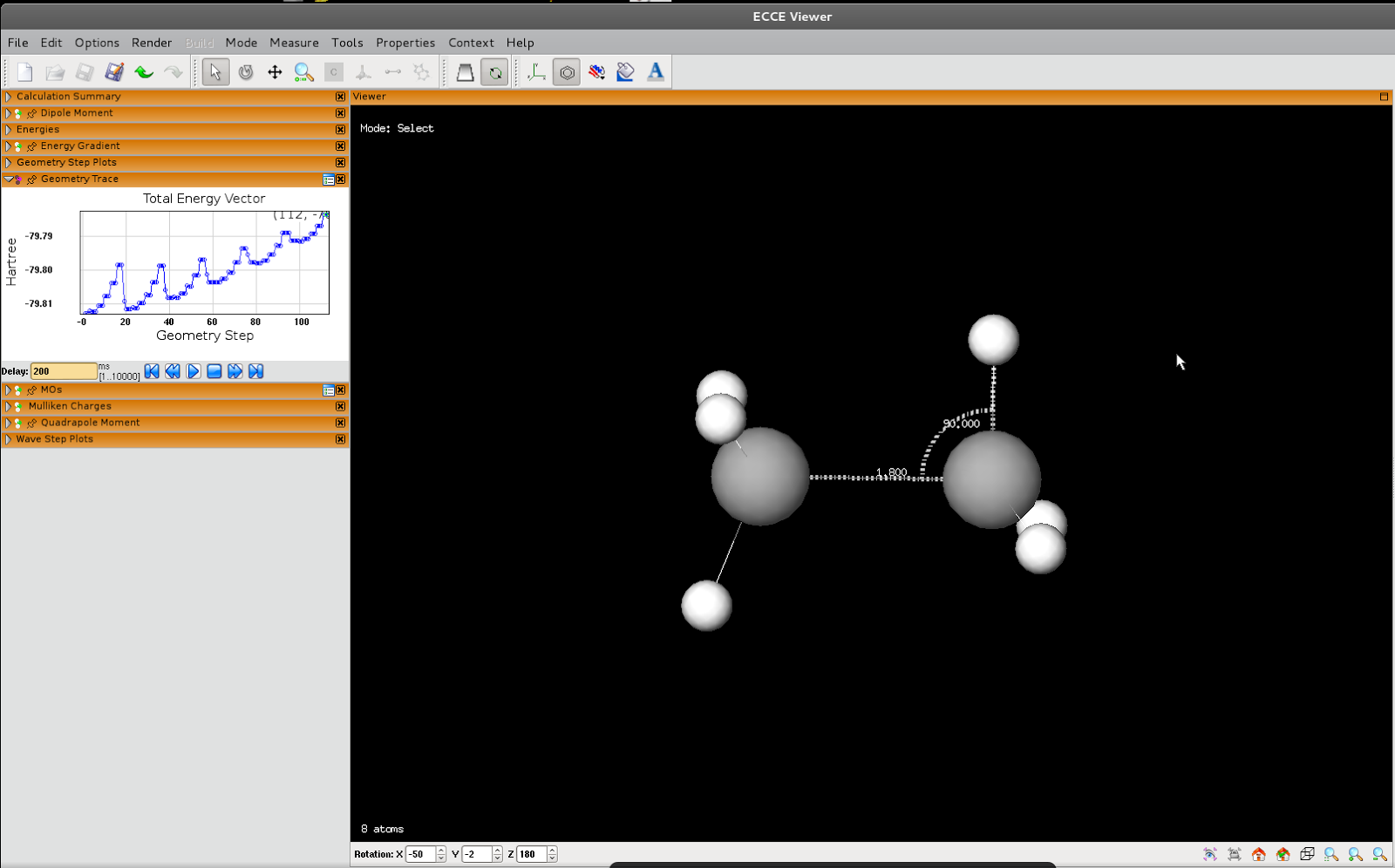
Two-dimensional PES scan
I'll keep this brief. First we do a scan where we use 'constant' for the angle, but not the bond length:
scratch_dir //scratch
Title "2d_pes-1"
Start 2d_pes-1
echo
charge 0
geometry noautosym units angstrom
C -2.51242e-66 1.67495e-66 -0.767732
H -0.722530 0.722530 -1.16548
H -0.264464 -0.986995 -1.16548
H 0.986995 0.264464 -1.16548
C 2.51242e-66 -2.51242e-66 0.767732
H 0.264464 0.986995 1.16548
H -0.986995 -0.264464 1.16548
H 0.722530 -0.722530 1.16548
end
ecce_print ecce.out
basis "ao basis" cartesian print
H library "6-31G"
C library "6-31G"
END
dft
mult 1
direct
XC b3lyp
grid fine
iterations 99
mulliken
end
driver
default
end
python
from pes_scan import pes_scan
geom = '''
geometry noprint adjust
zcoord
bond 1 5 %f cc
angle 2 1 5 %f hcc constant
end
end
'''
results = pes_scan(geom, \
[1.535, 111.269],
[1.800, 90],
5, 'dft', task_optimize)
end
task python
And the output:


What's happening is that the bond length ends up being the same no matter what we initially set it to
python
from pes_scan import pes_scan
geom = '''
geometry noprint adjust
zcoord
bond 1 5 %f cc constant
angle 2 1 5 %f hcc constant
end
end
'''
results = pes_scan(geom, \
[1.535, 111.269],
[1.800, 90],
5, 'dft', task_optimize)
end
task python
And we get:

This is dummy text. It is not meant to be read. Accordingly, it is difficult to figure out when to end it. But then, this is dummy text. It is not meant to be read. Period.
ConversionConversion EmoticonEmoticon